

- within Corporate/Commercial Law topic(s)
- with readers working within the Business & Consumer Services industries
- within Corporate/Commercial Law, Tax and Antitrust/Competition Law topic(s)
当前,跨境BD(Business Development)交易已成为中国创新药企实现研发资产变现、对接全球市场、分散商业化风险的核心路径,License Out、NewCo、合作开发、资产收购等各类BD模式,均普遍采用“首付款+里程碑付款+销售分成”的结构化对价安排,其本质是“风险与收益分层分配”的商事契约,许可方(Licensor)通过前期研发投入换取阶段性对价,被许可方(Licensee)则承担后续开发与商业化风险以博取长期收益。然而,在BD交易协议签订后的履约过程中,里程碑付款拒付、研发义务消极履行、被许可方单方终止项目等争议时有发生。
美国特拉华州作为全球医药BD交易常见的准据法及争议管辖地(包括中国药企作为BD交易一方的情况),其衡平法院判决以说理充分、逻辑严密著称,相关纠纷的裁判规则对全球医药BD交易的司法实践具有参考价值。2024年9月至2025年6月,特拉华州衡平法院就SRS(Syntimmune原股东代表)诉Alexion案先后发布了两份长篇意见书,分别就责任认定与损害赔偿作出裁判。该案涉及的交易虽形式上为股权并购,但其本质上仍为药品管线交易,且对价结构(4亿美元首付款+最高8亿美元里程碑付款)与典型的License‑out交易高度同构,法院对“里程碑达标认定”“商业合理努力义务履行”“预期损失量化”三大核心争议的说理,为采用结构化对价安排的BD交易合同提供了具有先例价值的裁判参照。
本文以该案判决为参照,从交易背景、合同条款、履约过程入手,重点分析法院对里程碑达标认定、商业合理努力义务履行标准、预期损失量化方法三大争议焦点的裁判逻辑,并从中提炼可供中国许可方参照的实务启示,助力中国创新药企在BD交易出海过程中规避风险、锁定核心权益。
01.案件背景与合同框架
(一)本案所涉交易概况
2018年9月,Alexion Pharmaceuticals, Inc.(下称“Alexion”)与Syntimmune, Inc.(下称“Syntimmune”)签订《合并协议》,Alexion收购Syntimmune及其核心管线——一款针对IgG自身免疫性疾病的抗FcRn单克隆抗体,交易前代号为SYNT001,交易后更名为ALXN1830。1 IgG是免疫球蛋白G(Immunoglobulin G),本案中ALXN1830靶向降低的抗体,其降低幅度是衡量药效的核心指标。根据判决书的说明,IgG是人体内最常见的有效抗体类型,在正常免疫反应中发挥抵御病原体的作用;但在IgG介导的自身免疫性疾病中,IgG因异常识别转而攻击健康细胞。FcRn是一种使IgG在血液中存留更长时间的蛋白质。抗FcRn单克隆抗体(如ALXN1830)通过与FcRn结合,阻断FcRn对IgG的保护作用,从而减少循环中的IgG数量,减轻对健康细胞的损伤。
《合并协议》规定的对价结构为2:
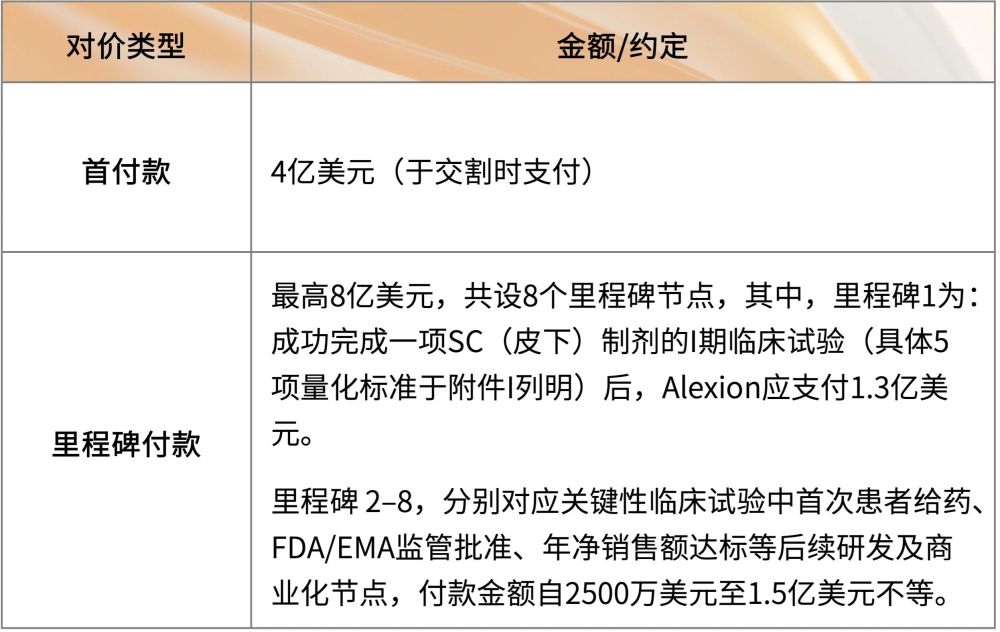
《合并协议》同时指定Shareholder Representative Services LLC(下称“SRS”)作为Syntimmune原全体股东的授权代表。合并于2018年11月完成。
(二)商业合理努力义务的设定
《合并协议》第3.8(f)条规定,Alexion在交割后7年内须以“商业合理努力”推进ALXN1830管线的研发与商业化,以实现各项里程碑。《合并协议》对商业合理努力的定义为外向型标准,即参照与Alexion规模、范围相似的生物制药公司,在开发类似阶段、类似产品时所通常投入的努力与资源,并考量产品的优劣势、市场竞争格局、监管批准可能性、预期盈利等商业与科学因素。3
(三)皮下制剂在交易中的重要意义及其他背景信息
判决书第27页记载,Alexion在尽职调查中即认为,ALXN1830要在竞争日益激烈的抗FcRn药物市场中取得成功,须“快速进入市场并与竞争对手形成差异化”,而实现差异化的最佳路径是“率先推出适用于低剂量皮下注射的制剂,可由患者通过体表给药装置等设备自行操作”。4 交割时,Syntimmune尚未在人体中测试皮下制剂,皮下制剂给药装置亦未选定,但Alexion仍将皮下制剂的开发前景作为评估交易价值的重要因素。5并且,《合并协议》里程碑1付款的触发条件围绕皮下制剂临床试验的推进,约定为以下较早发生者:(A) 通过满足附件I所列标准证明皮下制剂已完成I期临床试验;或(B) 向美国食品药品监督管理局(FDA)提交任何皮下制剂的关键性临床试验方案。6
在协议谈判过程中,双方围绕皮下制剂商业可行性的具体达成指标进行了多轮讨论。Alexion在2018年8月3日向Syntimmune发出的要约函中,以七项技术指标描述其对皮下制剂的商业预期,具体包括:单次给药体积不超过3.5 mL、浓度不低于150 mg/mL、给药频率每周一次或更低频次、与静脉注射制剂相当的半衰期/药效学/耐受性、维持足够药物暴露以维持IgG抑制作用、良好生物利用度、无免疫原性负面影响等。7
Syntimmune在收到上述提案后,进行了内部讨论并提出了反驳意见。经过多轮谈判,双方最终在《合并协议》附件I中确定了五项标准8,分别为:
1.观察到的PK/PD特征支持在长期安全性和有效性研究中进行每周一次或更低频次的皮下给药;
2.浓度为150 mg/mL的原料药和150 mg/mL瓶装制剂在预期储存条件下均能保持至少9个月的稳定性。;
3.稳态下总IgG平均降低50%,且IgA、IgM或白蛋白无有意义的变化;
4.安全性和耐受性特征允许在后续皮下制剂临床研究中继续对队列进行给药;
5.以总IgG降低为证明依据,抗药物抗体(Anti-drug antibody, ADA)数据未对PK/PD产生有意义的影响。
其中,关于PK/PD对本交易的意义,法院总结道,安全性与有效性亦可通过数据进行评估,临床研究者会监测药代动力学(Pharmacokinetics,PK),即药物进入、通过及排出体内的过程,以及评估药效学(Pharmacodynamics,PD),即药物对机体产生的效应。在本交易中,PK通过体内药物浓度进行衡量,而PD则通过IgG降低水平进行评估。9
这一演变过程体现了双方在商业预期与合同可执行性之间的博弈与妥协,最终形成的五项标准兼具客观性与可验证性,为后续里程碑1达成的认定奠定了清晰的合同基础。
02.履约过程
本案《合并协议》于2018年9月28日签署,同年11月2日正式交割。交割后,Alexion取得ALXN1830管线的控制权,并依据协议约定开展后续开发活动。根据判决意见书,详细履约过程如下:
(一)交割时的管线状态与买方知悉的风险信息
交割时,ALXN1830处于临床前末期向I期临床过渡阶段。卖方Syntimmune已完成SYNT‑101试验(在健康志愿者中开展的静脉制剂I期试验),结果显示耐受性良好,总IgG降低达到概念验证标准。SYNT‑102系在温抗体型自身免疫性溶血性贫血(Warm Autoimmune Hemolytic Anemia,WAIHA)患者中开展的静脉制剂1B/2A期试验;SYNT‑103系在寻常型天疱疮(Pemphigus Vulgaris,PV)患者中开展的静脉制剂1B/2A期试验。两项试验在交割时仍在进行中。皮下制剂尚未在人体中测试,皮下制剂给药装置亦未选定。由于按照当时的进展,静脉制剂在WAIHA适应症的开发进度比皮下制剂快了近两年,因此Alexion在交割后仍继续推进静脉制剂的开发,以尽早进入市场。
在尽职调查中,Syntimmune向Alexion披露了以下风险信息:静脉制剂曾出现输液相关反应(Infusion Related Reactions,IRR),均为1至2级的轻度不良事件,未报告严重不良事件;ALXN1830在健康志愿者及患者中的抗药物抗体(ADA)阳性率历史数据位于63%至91%区间,中和抗体(Neutralizing Antibodies, nAb)阳性率位于44%至65%区间。10 关于ADA和nAb的临床意义,判决意见指出:在某些情况下ADA可能降低治疗产品的疗效或在给药后引发不良事件,因此临床前及临床研究中需全程监测ADA的发生率。nAb是ADA的一个亚类,nAb可能会阻断药物的药理作用,从而降低药物的治疗效果,导致需要增加给药剂量。11
(二)I期临床阶段的推进与里程碑1相关数据的产生
交割后,Alexion继续推进静脉制剂的临床开发,但遭遇系列挫折:SYNT‑104(静脉制剂I期)因输液相关反应于2019年3月终止;SYNT‑102亦于同年晚些时候终止;FDA拒绝了Alexion在WAIHA适应症开展2/3期无缝试验的申请,理由是剂量‑反应、疗效和安全性数据不足,但未对所提交数据的安全性提出质疑。12 2020年春季,Alexion全面放弃静脉制剂开发,将资源集中于皮下制剂。
在皮下制剂方面,Alexion于2019年12月在英国启动HV‑105试验(皮下制剂首次人体I期),后因英国COVID‑19疫情暴发而暂停。2020年3月起,Alexion规划在新西兰开展HV‑108试验作为替代研究。HV‑108于2021年3月开始给药,为单次及多次剂量递增研究,共设6个剂量队列,13
2021年9月,Alexion收到HV‑108部分初步数据;2021年11月收到补充数据,包括队列3的完整PK/PD及免疫原性数据。数据显示:队列3中接受活性药物的6名受试者ADA及nAb阳性率均为100%;在12周给药期间内,总IgG水平持续降低;试验期间未报告严重不良事件。14
上述数据出具后,Alexion未支付里程碑1对应的1.3亿美元里程碑款项。
(三) COVID 19疫情对试验进程的影响
2020年初,COVID 19疫情对Alexion正在进行的临床试验产生了以下客观影响:
2020年1月31日:Alexion在英国开展HV 105及HV 106试验的合同研究组织因英国出现COVID 19病例,主动暂停了上述两项试验的给药。
2020年3月:Alexion研发团队基于对WAIHA患者群体在疫情期间的风险评估,建议暂停WAI 201试验,并获得Alexion研发领导层批准。至2020年3月底,Alexion所有ALXN1830相关临床试验均处于暂停状态。15
(四) 研发资源调整
2020年4月,Alexion对其研发优先级进行调整。Alexion在内部邮件中说明,公司需重新平衡研发组合以确保实现“在2023年前推出10款产品(10 by 2023)”目标。在此背景下,ALXN1830项目的研发预算被削减,部分资金被转移至其他优先级更高的项目。预算削减导致当2020年9月皮下制剂临床供应恢复可用时,HV 108试验的试验方案尚未完成撰写,试验启动因此延迟数月。16
(五)Alexion被收购与ALXN1830项目的最终终止
1. Alexion被收购
2021年7月,Alexion被另一家药企收购,双方拟通过收购承诺实现5亿美元的年度协同效应。收购完成后,Alexion对全部在研药物管线启动全面审查。17
2. 部分二期研究的暂停
在 Alexion被收购后的管线审查过程中,Alexion对ALXN1830项目的推进采取了以下措施:
2021年8月:Alexion暂停了MG 201研究的受试者筛选,这项研究是皮下制剂在全身型重症肌无力(generalized Myasthenia Gravis,gMG)患者中的二期试验;同时宣布ALXN1830项目将新增两种适应症甲状腺眼病(Thyroid Eye Disease,TED)和慢性抗体介导性排斥反应(chronic Antibody Mediated Rejection,cAMR)。
2021年9月:Alexion暂停了WAI 202研究(皮下制剂在WAIHA患者中的二期试验)。18
3. Alexion委托外部顾问对HV 108数据进行评估
2021年11月,Alexion委托免疫原性领域的外部顾问Paul Chamberlain对HV 108试验数据进行独立评估。Chamberlain于2021年11月23日出具分析报告,其中载明以下结论:
“基于HV 108研究中SC给药的结果,检测到的ADA应答似乎并未损害整体获益 风险平衡”;“有充分的证据权重支持恢复HV 108研究并推进至下一临床开发阶段”。19
4. 项目终止决策与通知
2021年12月14日,Alexion召开内部的罕见病早期开发审查委员会会议。会议纪要记载,委员会经讨论后决定终止ALXN1830项目的开发。会议纪要中列出的考量因素包括:免疫原性风险、药物累积现象、进入市场次序、以及COVID 19与动物死亡事件导致的开发延迟。
2022年1月,Alexion向SRS发出书面通知,正式告知ALXN1830项目终止。通知函中列明的终止理由包括:ALXN1830的药代动力学特性局限、较大的给药体积与装置挑战、不利的免疫原性特征、以及尚无法解释的免疫原性与药物累积问题。20
5. HV 108临床研究报告的发布与修订
2022年8月,Alexion发布HV 108试验的临床研究报告。报告原文载明:“总体而言,在各剂量组中,无论是否存在ADA、nAb或高ADA滴度,均未观察到对药物浓度(PK特征)或IgG(PD指标)水平的影响。”
2023年5月,在本案诉讼进行期间,Alexion发布了HV 108试验临床研究报告的修订版本。修订版本将上述语句修改为:“总体而言,在各剂量组中,无论是否存在ADA、nAb或高ADA滴度,均未观察到药物浓度(PK特征)或IgG(PD指标)水平的降低。”即,将“no impact”改为“no reduction”。21
03.争议焦点与裁判规则
特拉华州衡平法院对本案的审理历时数年,先后于2024年9月和2025年6月发布了两份长篇《意见书》,分别就责任认定与损害赔偿计算作出裁判。以下结合判决书内容,对三项核心争议焦点的法院说理与裁判规则进行详细拆解。
(一)里程碑1达成的认定
1. 双方的争议焦点
SRS主张,HV‑108试验数据已满足《合并协议》附件I就里程碑1所列全部五项标准,里程碑1已于2021年11月达成,Alexion应支付1.3亿美元里程碑款项。Alexion则抗辩称,标准1要求HV‑108数据不仅支持每周或更低频次给药,还须支持ALXN1830在长期安全性与有效性研究中的实际给药;由于HV‑108数据显示出无法解释的药物累积现象,需进一步研究后方可开展长期研究,故里程碑1尚未满足。此外,Alexion在项目终止决策中亦质疑了ALXN1830的安全性与商业可行性。22
2. 法院的合同解释
法院对附件I的五项标准进行了解释,其核心裁判逻辑如下:
第一,争议范围仅限于标准1和5。法院首先确认,Alexion在诉讼中已自认标准2(浓度与稳定性)、标准3(IgG降低幅度)、标准4(安全性与耐受性)均已满足,双方仅对标准1和5的解释与满足状态存在争议。后续法院重点对标准1和5进行分析。23
第二,标准1的文义存在歧义,但外部证据支持SRS的解释。标准1的原文为:“An observed PK/PD profile that supports weekly or less frequent subcutaneous administration in long term safety and efficacy studies.”(观察到的药代动力学/药效学谱支持在长期安全性和有效性研究中进行每周一次或更低频次的皮下给药。)法院认定该条款的文义可同时支持双方的合理理解,构成歧义。但基于谈判历史等外部证据,法院最终采纳了SRS的解释:标准1仅要求数据支持每周或更低频次给药,不要求数据证明可立即开展长期研究。24
第三,标准5的衡量标准是唯一且明确的。标准5涉及的是抗药物抗体(ADA)对药效的影响。针对双方争议最大的标准5,法院认定其文义“清晰无歧义地提供了一项衡量指标:IgG降低”。换言之,判断ADA是否对药物产生有意义影响,唯一的标准是观察IgG的降低幅度是否受到影响,而无需考量ADA阳性率高低、药物累积现象或其他免疫原性指标。尽管IgG降低未必是评估ADA对药代动力学(PK)影响的“最直接”指标,但“当事人有权订立好的合同与坏的合同,法律均予执行”,法院无权以事后视角替当事人修改合同条款。25
第四,Alexion的抗辩与客观证据矛盾。法院采信了HV‑108原始临床研究报告、Alexion内部会议记录及外部顾问Chamberlain报告的结论,认定尽管ADA及nAb阳性率达100%,但IgG降低未受有意义影响,标准5已满足。Alexion在诉讼期间对HV‑108临床研究报告的修订,法院明确“不予采信”。26
(二)商业合理努力义务的违反认定
1. 双方的争议焦点
SRS主张,Alexion在里程碑1触发后大幅削减研发预算、缩减团队、并最终终止项目,未按约定履行商业合理努力义务。Alexion则抗辩称,其终止项目系基于“战略调整”及Alexion被收购后的“协同效应考量”,属于商业判断自由,应豁免其商业合理努力义务;且其已投入部分资金,应认定已履行合理努力。
2. 法院对CRE条款的解释
《合并协议》第3.8(f)条对CRE的定义采用了外向型标准,要求参照“同类规模生物医药公司在类似开发阶段对类似产品的典型努力”,原文为:
“在评估[SYNT001]的研发及商业化进程时,需综合考量以下因素:与[Alexion]规模及业务范围相近的生物制药企业通常采用的资源投入策略;[SYNT001]的优劣势特征、疗效表现、安全性数据、监管部门批准的药品标签与定价方案、市场竞争力、孤儿药资格认定、专利保护范围及技术专有性地位、研发成功率与监管审批概率、涉及的监管架构、预期盈利潜力,以及其他同类产品开发过程中生物制药企业普遍考量的科技参数与商业要素。” 27
3. 法院在解释该条款时,确立了以下裁判规则:
A. 并购协议中约定的商业合理努力义务采取了外向型标准:
Alexion的努力程度应以理性第三方同类企业的典型行为为参照,而非其自身的内部战略优先级或财务压力。法院在判决书明确指出,商业合理努力条款“界定了当事人为实现目标必须付出的努力程度。”该条款具有外向性特征,意味着其设定的是客观标准。对于具有外向性特征的条款而言,Alexion公司的“主观意图或心理状态”并不能决定其是否履行了商业合理努力义务。Alexion承诺将采用一家与自身规模相当的假设典型公司在开发类似ALXN1830分子时所投入的努力和资源,该评估基于此类公司通常会考量的因素,直至出现违背审慎商业判断的情形。该条款旨在根据Alexion的资本状况、ALXN1830的研发进展以及分子特性演变进行动态调整。该基准并非固定不变,需通过事实调查来界定Alexion在ALXN1830研发历程中任一时间节点的基准值。28
B. 在外向型标准下,“内部战略调整”不构成免责事由:
Alexion以“10 by 2023”内部战略及被收购后追求并购协同效应为由削减投入、终止项目,并非基于常规考量因素,而是基于企业特有的举措,不属于法定的或约定的免责理由,未能达到商业层面上合理的投入标准。29
C. 参考行业对标公司的研发情况和企业内部评价
本案中,双方确定了四家Alexion的竞争对手,对比分析了Alexion的研发进展及其产品表现,这四家公司均从事抗FcRn疗法的生物制药研发。所有竞争对手的规模在当时均未显著超过Alexion,且均未拥有实质性的资源优势。Alexion公司在2020年对ALXN1830的研发项目进行降级处理,其投入力度远未达到商业层面可接受的标准。若以Alexion为参照案例,综合考量此类企业通常考量的因素,任何类似公司都不会对这类产品进行如此大幅度的资金削减。2020年Alexion的竞争对手们都在推进类似产品的研发。所有竞争对手在当年都对其特定分子的临床开发取得了一定进展。Alexion公司本身也将ALXN1830项目视为高优先级项目和关键战略重点,根据Alexion内部观点,认为ALXN 1830的研发进程需要加速推进而非放缓。30
法院据此认定,Alexion的努力程度“远低于行业常规水平”,未达到商业合理努力义务的履行标准。31 同时,法院穿透审查了Alexion的终止动机,认定现有证据表明其决策受到其被收购后追求并购协同效应的实质性影响与驱动。32
(三)预期损失的量化
1. 损害赔偿计算的独立审理
本案的损害赔偿问题并未在2024年9月的责任认定意见书中处理。2025年6月11日,法院发布第二份《备忘录意见书》,专门就Alexion违反商业合理努力义务所导致的预期损失进行了量化裁判。
2. 预期价值法的采用
法院认为,SRS因Alexion违约所遭受的损失,并非“确定会获得的里程碑款项”,而是“获得或有对价的合同权利的期望值减损”。法院在第二份意见书中写道:SRS的损失最适宜理解为各里程碑事件在Alexion违反其商业合理努力义务前后预期价值的差异。当原告能够证明被告的违约行为是其损失的事实原因(but-for cause)时,预期损害赔偿金当然具有可获得性。本案中,Alexion公司的违约行为是终止ALXN1830项目,该行为导致违约时所有里程碑指标的预期价值完全消失。这种终止行为正是SRS公司预期价值损失的事实原因:由于缺乏时间或机会让其他间接因素介入,导致其预期价值彻底丧失。法院并非认为SRS的损失为全部里程碑付款的损失整体,但法院支持SRS的损失了其对里程碑付款的预期价值。33
特拉华州法律赋予法院灵活性,允许其合理确定此类估算值。
法院采用了预期价值法,计算方式为:
预期损失 = Σ(各里程碑约定金额×该里程碑在违约时的实现概率)折现至违约日的现值
3. 实现概率的认定
在认定各里程碑的实现概率时,法院综合考量了以下数据:
A. Alexion内部的评估数据:Alexion在收到HV 108数据前,内部评估cAMR适应症的技术与监管成功概率(probability of technical and regulatory success,PTRS)为34%,TED适应症的PTRS为30%。34 法院认为该数据系Alexion管理层基于对管线的深入了解所作出的商业判断,且未受终止意图的影响,具有较强的证明力。Alexion在收到HV 108数据后将两个适应症的PTRS均大幅下调至10%的行为,因缺乏科学依据且与内部评估矛盾,未被法院采信。35
B. 行业平均成功概率:SRS的专家证人Kinch基于其自行建立的实验药物临床经验知识库对部分里程碑的概率进行了预估。36 法院对该知识库的可靠性表示保留(因其仅包含公开数据,可能系统性高估成功率),但未完全排除其参考价值。37
C. 合同约定的里程碑触发条件:法院根据协议中里程碑 2至里程碑 8的具体约定,逐项计算了各里程碑的预期实现概率,并运用概率论公式(条件概率、独立事件概率、全概率公式)进行了详细推导。
4. 折现率的选择
法院对不同类型的里程碑适用了不同的折现率,并就其选择折现率的理由进行了论述38:
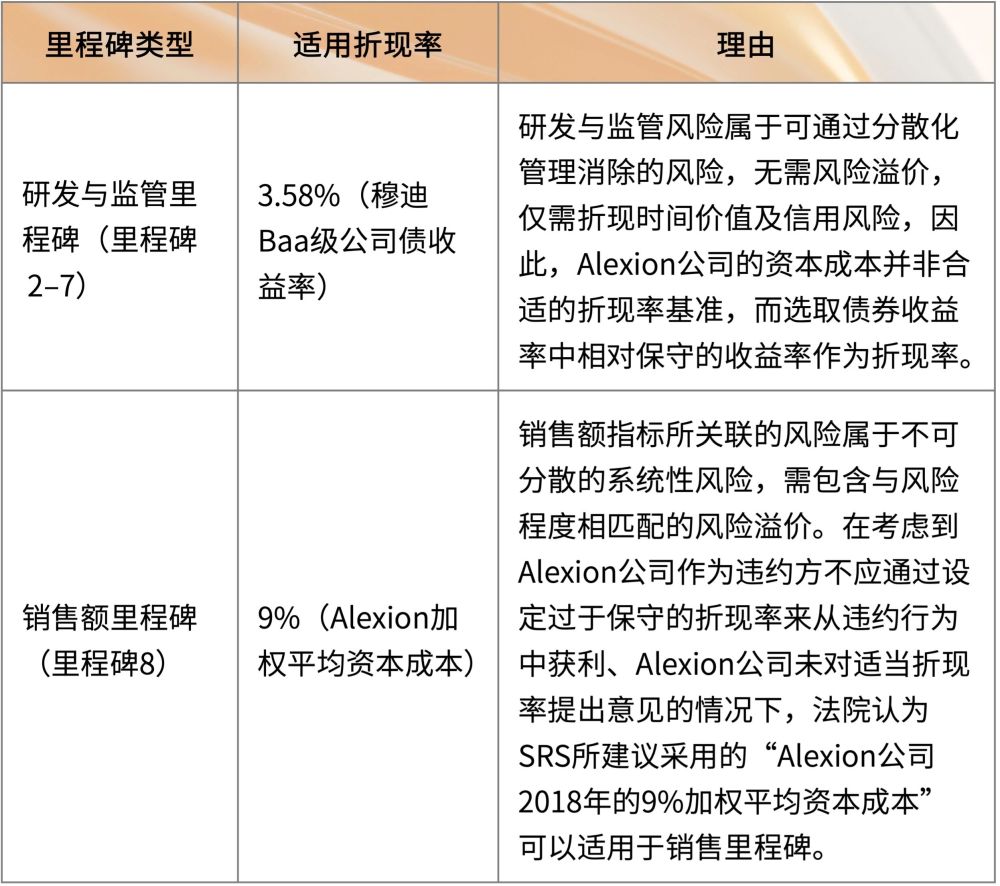
5. 最终赔偿金额
法院经上述计算,最终判令Alexion就违反商业合理努力义务向SRS支付预期损害赔偿 180,944,915.32美元(约1.81亿美元),外加判决前后利息。39
04.对中国创新药企作为许可方的法律实务启示
本案中,Syntimmune在合同设计、履约管理和证据留存上的充分准备,是其赢得诉讼和挽回损失的关键所在,为中国创新药企在BD交易的谈判和推进过程提供了可借鉴的实务启示:
1.里程碑条款的起草:法律团队与科学团队应密切协作,以尽可能明确里程碑的具体达成标准
Alexion案中,对于里程碑1的具体达成标准,双方经过多轮讨论,并最终确定5项较为客观的细分指标,为法院审理时提供了相对明确的参照。但法院认为由于合并协议中里程碑的表述存在歧义,因此对外部证据进行了全面审查。如果起草时的表述本身更为清晰,本可避免这一情况。此外,值得注意的是,在双方对里程碑1的具体达成标准进行讨论的过程中,Syntimmune的员工曾评价该Alexion起草的条款“看起来像是律师试图定义自己都不理解的事物”40。在协商和起草里程碑条款时,法律团队应与其科学和业务对应方密切协作,确保条款起草准确无误,并考虑可能存在的其他解释,以避免代价高昂的诉讼及潜在的不可预测结果。
2.商业合理努力的起草:注意区分“外向型”与“内向型”标准
“内向型”标准通常可能更有利于被许可方/买方,因为其允许考量被许可方/买方自身的做法和立场,该标准下的要求可能因双方约定的条款而存在显著差异,而这些条款往往技术性极强,且需经过多轮协商。在起草商业合理努力条款时,应考量“外向型”与“内向型”标准的优劣,同时还需斟酌任何定制化条款——这些条款需明确买方在实现业绩承诺里程碑时可参考的相关标准,以及被许可方/买方拥有的裁量权。无论采用哪种标准,在该领域都应审慎预判约定条款如何适用于实际商业运营。从许可方/卖方的角度,“外向型”标准因为要求被许可方/买方以“同类规模企业在类似阶段对类似产品的典型努力”为参照,而非以被许可方/买方自身的内部优先级或历史投入水平为衡量尺度,通常更有利于许可方/卖方。
以下为可供参考的“外向型”商业合理努力条款示范文本,中国许可方/卖方可结合具体交易情况,在协商中争取纳入类似表述:
“Commercially Reasonable Efforts” means, with respect to the Product, using such efforts and resources typically used by biopharmaceutical companies similar in size and scope to [Licensee/Buyer] for the development and commercialization of similar products at similar development stages taking into account, as applicable, the Product’s advantages and disadvantages, efficacy, safety, regulatory authority-approved labeling and pricing, the competitiveness in the marketplace, the status as an orphan product (if applicable), the patent coverage and proprietary position of the Product, the likelihood of development success or Regulatory Approval, the regulatory structure involved, the anticipated profitability of the Product, and other relevant scientific, technical and commercial factors typically considered by biopharmaceutical companies similar in size and scope to [Licensee/Buyer] in connection with such similar products.
“商业合理努力”指在产品开发过程中,与[被许可方/买方]规模及业务范围相当的生物制药企业为开发和商业化类似产品所通常采用的努力与资源投入。该评估需综合考量以下因素:产品的优劣势、疗效、安全性、监管部门批准的标签与定价、市场竞争力、孤儿药资格(如适用)、专利保护范围及产品专有地位、研发成功率或监管审批可能性、涉及的监管框架、产品预期盈利能力,以及其他与规模相当企业评估类似产品时通常考量的科学、技术及商业因素。
3.重视谈判期间的同期记录:
在对里程碑的含义进行解释时,法院从最初的Alexion提案开始,详细审查了谈判历史,同时也审查了体现Alexion自身对相关条款同期解读的内部沟通记录。这一案例警示相关方,书面记录(包括内部电子邮件)可用于解读一方当事人的真实意图。尤其是某些内部沟通若存在“唱反调”的情况,且未明确说明这一立场时,可能会被用作对该方不利的证据,佐证对模糊条款的不利解读。因此,考虑到证据开示规则的适用,交易各方即便在内部讨论中,也应重视其所形成的书面记录。
05.结语
SRS v. Alexion案作为特拉华州衡平法院近年来审理的最具代表性的跨境BD合同纠纷之一,其裁判文书对里程碑客观达标、CRE外向型标准、预期价值损害赔偿三大核心问题作出了详尽、系统的说理,为同类交易的风险防控与争议解决提供了宝贵的司法指引。对于中国创新药企而言,在License out/BD交易中主动借鉴本案的裁判逻辑,从条款设计、履约监管、证据储备三个维度提前布局,将有效降低出海过程中的法律风险,切实维护自身在里程碑付款及销售分成中的合法权益。
【注:本文初稿由周迎春执笔完成】
文章附录
脚注:
1 2024年责任认定意见书(下称“2024 Op.”)第1页。
2 2024 Op.第37页,2025年损害赔偿意见书(下称“2025 Op.”)第3页。
3 2024 Op.第38–39页。
4 2024 Op.第27页。
5 2024 Op.第40页。
6 2024 Op.第37–38页。
7 2024 Op.第30页。
8 2024 Op.第24页。
9 2024 Op.第11页。
10 2024 Op.第41-42, 60页。
11 2024 Op.第12页。
12 2024 Op.第41–44页。
13 2024 Op.第45–46页。
14 2024 Op.第59,67,122页。
15 2024 Op.第46–49页。
16 2024 Op.第50–53页。
17 2024 Op.第57–58页。
18 2024 Op.第58,63页。
19 2024 Op.第69-71页。
20 2024 Op.第77–80页。
21 2024 Op.第80–81页。
22 2024 Op.第85页。
23 2024 Op.第84–88页。
24 2024 Op.第90-96页。
25 2024 Op.第99-101页。
26 2024 Op.批注569。
27 2024 Op.第38页。
28 2024 Op.第107、109、112页。
29 2024 Op.第112-116页。
30 2024 Op.第113-116页。
31 2024 Op.第113页。
32 2024 Op.第137页。
33 2025 Op.第18页。
34 2025 Op.第28页。
35 2025 Op.第50-51页。
36 2025 Op.第10-12页。
37 2025 Op.第48页。
38 2025 Op.第54-59页。
39 2025 Op.第59-64页。
40 2024 Op.第35页。
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.
[View Source]
